
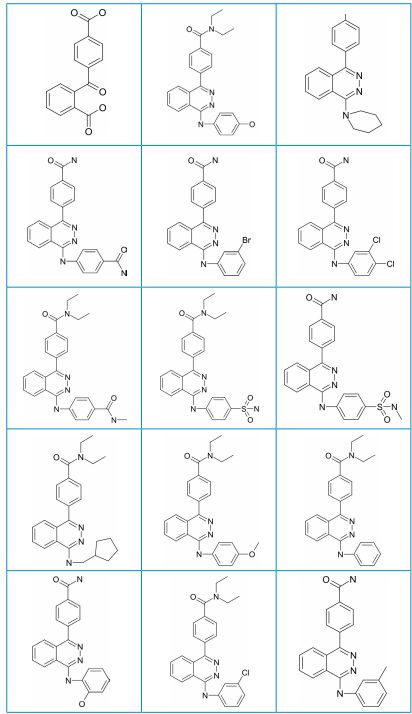
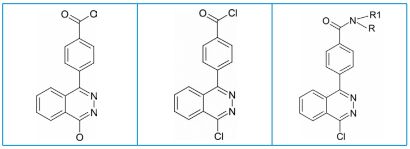
Синтез и изучение биологической активности фталазинкарбоновых кислотУДК 576.89 Оригинальное эмпирическое исследование Зубенко А. А., Фетисов Л. Н., Святогорова А. Е., Яровая Н. А., Байрамова В.Н. Северо-Кавказский зональный Аннотация. Конденсированные диаза-гетероциклы составляют основу многих биоактивных природных продуктов и эффективных терапевтических препаратов. Среди них фталазины признаны выдающимися структурными лидерами в медицинской химии благодаря их широкому применению в фармацевтической и агрохимической промышленности. Получение доступа к таким сложным фармацевтическим агентам с высокой химической сложностью с помощью синтетически эффективных подходов остается привлекательной целью в современной медицинской химии и в области разработки лекарств. Целью исследований являлся синтез и изучение биологической активности новых производных фталазинкарбоновых кислот. Ключевым соединением является дикарбоновая кислота №1 взаимодействием которой с гидразингидратом получена фталазиновая кислота А. Реакцией фталазиновой кислоты А с хлорокисью фосфора получали дихлорпродукт Б, который в мягких условиях реагирует с соответствующими аминами с образованием амидов В. Реакцией амидов с соответствующими нуклеофилами при кипячении в метилцеллозольве получено 15 новых соединений. Методами, принятыми в экспериментальной химиотерапии, изучали биологическую активность новых соединений. Было установлено, что большинство соединений обладают активностью в той или иной степени. Значимый уровень антибактериальной активности обнаружен у дикислоты № 1 в отношении Staphylococcus aureus, равный уровню активности препарата сравнения фуразолидона, а также заметная активность в отношении Esherichia coli. Представляется перспективным синтез аналогов данной дикислоты как с донорными, так и акцепторными заместителями с целью усиления активности. Также выявили у соединения № 14 значительную антипротозой-ную активность, равную уровню активности препарата сравнения толтразурила. Значительная фунгистатическая активность наблюдается у соединения № 10 с липофильным заместителем как в отношении Penicillium italicum, так и Staphylococcus aureus. Введение липофильных заместителей может стать перспективным направлением модификации структур данного ряда по усилению биологической активности. Ключевые слова: фталазин, фталазинкарбоновые кислоты, биологическая активность, антипротозойная активность, фунгистатическая активность, антибактериальная активность Конденсированные диаза-гетероциклы составляют основу многих биоактивных природных продуктов и эффективных терапевтических препаратов. Среди них фталазины признаны выдающимися структурными лидерами в медицинской химии благодаря их широкому применению в фармацевтической и агрохимической промышленности. Получение доступа к таким сложным фармацевтическим агентам с помощью синтетически эффективных подходов остается привлекательной целью в современной медицинской химии и в области разработки лекарств. Sumera Zaib и Imtiaz Khan (2020) сосредоточились на последних разработках в области синтеза активных фармацевтических ингредиентов на основе фталазина и их биологическом потенциале в отношении различных мишеней. Подробное изучение взаимосвязи структуры и активности фталазинового ядра, которое стало возможным благодаря замене фармакофора, выявило создание надежных, эффективных и более селективных соединений с выраженными биологическими эффектами. Таким образом, открытие новых структур-лидеров обещает улучшить методы лечения различных заболеваний, таких как туберкулез, лейшманиоз, малярия, болезнь Шагаса, а также многих других, включая различные виды рака, атеросклероз, ВИЧ, воспалительные и сердечно-сосудистые заболевания [3, 6, 12, 16]. Хорошо известно, что производные фталазинона представляют большой интерес в связи с их противодиабетическими, сосудорасширяющими, противоаллергическими, противоастматическими и гербицидными свойствами. Молекулы лекарственных средств, такие как гидралазин, бурдралазин, поналрест, азеластин и зополре-стат также являются производными фталазина. Было обнаружено, что несколько производных фталазина обладают противоопухолевым, противосудорожным, гипотензивным, антитрипаносомаль-ным, противомикробным и противовоспалительным действием. Ahmed T.A. Boraei et all (2019) отметили, что большинство лекарственных средств, содержащих фталазины, замещены во 2-м и 4-м положениях структуры фталазина, поэтому эта статья посвящена вариациям заместителей в этих двух положениях для поиска эффективных противоопухолевых препаратов [2]. Yurong Yang et all (2025) установили, что производные фталазина могут выступать как мощные модуляторы сборки капсидов вируса гепатита В. Капсид-белок вируса гепатита В образует защитный нуклеокапсид, необходимый для репликации вируса, что делает модуляцию сборки капсида многообещающей терапевтической стратегией, проявляя при этом субмикромолярную противовирусную активность in vitro. Путем химической модификации известных производных фталазина авторы усилили активность ряда соединений при одновременном повышении биодоступности в организме. Эти результаты подтверждают, что противоинфекционные средства на основе фталазина являются перспективными для дальнейшей разработки в качестве средств борьбы с гепатитом [18]. Азотсодержащие гетероциклы, содержащие рядом два атома азота, являются важным классом соединений благодаря их присутствию в природных и синтетических продуктах, которые проявляют полезную биологическую активность. Такие соединения представляют интерес благодаря своим фармакологическим свойствам и клиническому применению. Азотсодержащие гетероциклы с фталазиновым каркасом, такие как производные 1-арилами-но-2,3-дигидро-1Н-пиразоло[1,2-b]фталазин-5,10-диона, обладают противовоспалительными, обезболивающими, антигипоксиче-скими и жаропонижающими свойствами. Sajjad Keshipour et all (2012) разработали оригинальный метод синтеза фталазинкарбоновых кислот. Применение этого метода в синтетической и медицинской химии может быть значительным, поскольку продукты обладают сходными структурными и функциональными групповыми свойствами биологически активных молекул [14]. Усилия по разработке новых противоопухолевых средств в настоящее время направлены на разработку многоцелевых методов лечения, которые, как полагают, обладают высокой эффективностью и низкой склонностью к резистентности по сравнению с традиционными лекарствами. Kamilia M. Amin et all (2016) исследовали синтез и противоопухолевую активность пяти серий соединений на основе фталазина, содержащих различные биологически активные химические фрагменты в положении 1 фталазинового ядра. Противоопухолевая активность целевых соединений была проверена на четырнадцати линиях раковых клеток, причем все соединения были активны на наномолярном уровне. Кроме того, механизм действия целевых соединений был исследован с помощью анализа ферментативного ингибирования киназ VEGFR-2 и EGFR, который выявил мощную активность особенно по отношению к VEGFR-2. Способ связывания наиболее активных соединений был изучен с помощью докинг-экспе-римента [7]. Серия новых 2(4-алкоксифенил)циклопропилгидразидпроиз-водных и триазоло-производных была синтезирована Prithwiraj De et all (2010) на основе 4-гидроксикоричной кислоты по эффективной и простой технологии синтеза. Эти соединения были проверены на противоопухолевую активность in vitro в отношении 4 клеточных линий, обладающих определенным уровнем устойчивости к проапоптотическим механизмам действия, а также двухклеточных линий, чувствительных к проапоптотическим соединениям. Два соединения были наиболее активными и проявляли умеренный цитостатический эффект in vitro за счет различных механизмов. Важно отметить, что химически модифицированные производные могли быть получены для разработки новых типов соединений, направленных на борьбу с устойчивыми к апоптозу видами рака, например, с теми видами рака, которые ассоциируются с неблагоприятными прогнозами [13]. Бензодиазины (фталазин, хиназолин, хиноксалин и циннолин) стали привлекательной основой для создания новых противоопухолевых препаратов. Эти азотсодержащие гетероциклы интересны тем, что они имеют различные конфигурации и могут изменяться химически, что позволяет адаптировать их фармакокинетические и фармакодинамические характеристики. Многочисленные исследования показали, что производные этих соединений обладают мощными противоопухолевыми свойствами благодаря ингибированию топоизомераз, протеинкиназ и рецепторных тирозинкиназ. Эти соединения нарушают важнейшие процессы, которые контролируют распространение рака и выживаемость. Большинство производных бензодиазина успешно применяются в клиниках, продемонстрировав тем самым терапевтический потенциал гетероциклов. Использование производных фталазина, циннолина и хиназолина должно открыть новые возможности в разработке более эффективных и целенаправленных методов лечения рака. Mohamed S. Nafie et all (2025) обобщили последние достижения в синтезе этих соединений и показали, как они стали перспективными химиотерапевтическими средствами [10]. Mosaad S.M. Abd alla et all (2010) синтезировали и идентифицировали с помощью ИК, 1Н ЯМР, МС и элементного анализа некоторые новые производные 1,3,4-триазоло-, 1,3,4-оксадиазоло-, 1,3,4-тиадиазол- и пиразоло-3,4-диметилфенил-1(2Н)-оксофта-лазина. Большинство новых синтезированных препаратов были протестированы на их противовоспалительную активность. Среди них 5 соединений обладают активностью, сравнимой с активностью индометацина [11]. Серия 4-фталазинилгидразонов в Е-конфигурации продемонстрировала отличные противотрипаносомные и антилейшма-ниевые свойства in vitro. Предварительные анализы на обоих паразитах показали, что наиболее активные производные действуют через механизмы окислительного и нитрозативного стресса, однако точный механизм их действия в качестве антитрипано-сомальных и лейшманиозных агентов до конца не выяснен. Это побудило Angel H. Romero et all (2017) к проведению молекулярного исследования основных ферментов трипаносоматид, таких как супероксиддисмутаза, трипанотионредуктаза, цистеин-протеаза и птеридинредуктаза 1 [4]. Walid Ettahiri et all (2025) отметили, что среди широкого спектра гетероциклических соединений особое место занимают фталазин и его производные. Фталазин имеет бициклическую структуру, которая сочетает в себе бензольное кольцо с пиридази-новым кольцом. Такое уникальное расположение обеспечивает жесткую и стабильную структуру, которую можно легко модифицировать для повышения биологической активности. Слияние этих двух ароматических систем не только стабилизирует молекулу, но и создает возможности для разнообразных взаимодействий с биологическими рецепторами. Эти структурные особенности привлекли значительное внимание к производным фталазина в фармацевтических исследованиях. Производные фталазина продемонстрировали значительный потенциал в различных терапевтических областях. Они широко изучались как противоопухолевые, антиоксидантные, противомикробные, противовоспалительные, противогрибковые, противотуберкулезные, инсектицидные, анти-гербицидные, противодиабетические средства и средства против ВИЧ. Способность этих соединений поражать множество биологических мишеней делает их универсальными кандидатами для лечения широкого спектра проблем со здоровьем. Адаптивность производных фталазина и их способность поражать множество мишеней делают их многообещающими кандидатами при поиске новых лекарств против сложных заболеваний [17]. Sara M. Emam et all (2025) синтезировали производные фталазинкарбоновых кислот из исходного метил-3-(2-(4-бензил-1-оксофталазин-2(1Н)-ил) ацетамидо)пропаноата. Три соединения продемонстрировали значительную цитотоксичность, при этом значения IC50 составили 1,33, 0,41 и 0,38 мкм по сравнению с сорафенибом (IC50 = 2,93 мкм). Соединения 6d проявляли мощное ингибирование VEGFR2 на 97,6% при значении IC50 21,9 мкм по сравнению с сорафенибом (94,7% и значение IC50 30,1 мкм [15]. Mai M. Khalaf et all (2024) использовали простой и легко синтезируемый лиганд, а именно 1,4-диоксо-3,4-дигидрофтала-зин-2(1Н)-карботиоамид, в сочетании с 8-гидроксихинолином для создания новых комплексных соединений со смешанным лигандом, таких как Co (II) (C1), Ni (II) (C2) и Cu (II) (C3). Недавние исследования были посвящены изучению их антибактериальной, противогрибковой и противовоспалительной эффективности. Полученные комплексные соединения продемонстрировали значительно улучшенную антибактериальную и противогрибковую эффективность по сравнению со свободными лигандами. Кроме того, были исследованы противовоспалительные эффекты, используя метод денатурации яичного альбумина. Комплексы С1, С2 и С3 продемонстрировали значительное противовоспалительное действие [8]. Гидроксиапатит рыбьей чешуи был использован Cinnathambi Subramani Maheswari et all (2021) в качестве эффективного катализатора для синтеза производных 3-амино-1Н-пиразоло[1,2-b] фталазин-2-карбоксамида с помощью однокомпонентных реакций циклоконденсации ароматических альдегидов с цианоацетамидом и фталгидразидом в условиях, не содержащих растворителей. Синтезированные соединения были оценены на предмет их антибактериальной активности in vitro в отношении грамположи-тельных (Bacillus cereus, Bacillus aryabhattai, Bacillus megaterium, Staphylococcus aureus) и грамотрицательных (Serratia marcescens, Bacillus Pseudomonas putida, Escherichia coli, Enterobacter cloacae) бактерий. В этом исследовании в качестве стандартных антибиотиков использовались тетрациклин и эритромицин. В настоящем исследовании авторы добились значительных выходов продукта за более короткое время реакции, а катализатор можно было легко извлекать и использовать повторно [5]. Mohamed S. Behalo et all (2017) синтезировали серию производных фталазина в результате взаимодействия 1-хлор-4-(4-фе-ноксифенил)фталазина в качестве реакционноспособного исходного материала с различными углеродными, азотными, кислородными и сернистыми нуклеофилами. Некоторые из синтезированных производных были подвергнуты скринингу на их противоопухолевую активность в отношении четырех линий опухолевых клеток человека с помощью микротитрационного анализа (МТТ-тест). По сравнению со стандартным препаратом доксорубицином три соединения проявили наиболее выраженный цитотоксический эффект. Кроме того, исследование антиоксидантной активности показало, что гидразинилфталазин обладает наибольшей активностью [9]. Abdel-Ghany A. El-Helby et all (2020) отметили, что фталазин является привилегированной структурой и одним из наиболее перспективных классов гетероциклов, который хорошо переносится человеком и обладает противоопухолевой активностью. В результате проведенного авторами обзора литературы было обнаружено, что предпринималось много попыток изучить фталазиновый фрагмент как потенциальный интеркалятор ДНК с основной целью получения новых эффективных противоопухолевых средств. Основываясь на предыдущих своих результатах, авторы синтезировали три группы производных фталазина с целью получения многоцелевых молекул, обладающих активностью, направленной на ДНК и Topo II. Синтезированные соединения были разработаны таким образом, чтобы они обладали основными фармакофорными свойствами ДНК-интеркаляторов и ядов Topo II и могли таким образом обладать потенциально высокой антипролиферативной активностью. Синтезированные соединения ряда фталазина, содержащие два остатка карбоновых кислот были оценены in vitro на предмет их цитотоксической активности в отношении клеточных линий HepG-2, MCF-7 и HCT-116. Кроме того, для наиболее активных соединений была исследована ингибирующая активность Topo II и сродство к интеркаляции ДНК в качестве потенциального механизма противоопухолевой активности. Пять соединений проявляли наибольшую активность в отношении Topo II с IC50 в диапазоне от 5,44 до 8,90 мкм. Кроме того, одно соединение продемонстрировало in vivo значительное ингибирование роста опухоли [1]. Целью наших исследований является синтез и изучение биологической активности новых производных фталазинкарбоновых кислот. Как видно из представленного выше краткого литературного обзора, сведения о таких структурах довольно скудны (Sajjad Keshipour et all, Prithwiraj De et all, Sara M. Emam et all). Материалы и методы исследования. Химическая часть. Структурные формулы и номера синтезированных соединений представлены в таблице 1. Ключевым соединением является дикарбоновая кислота 1 (табл. 1), взаимодействием которой с гидразингидратом получена фталазиновая кислота А (табл. 2), реакция которой с хлорокисью фосфора приводит к дихлорпродукту Б, который в мягких условиях реагирует с соответствующими аминами с образованием амидов В. Реакцией В с соответствующими нуклеофилами при кипячении в метилцеллозольве получены соединения 2-15 (табл. 1). Таблица 1 Структурные формулы синтезированных соединений
Таблица 2 Ключевые исходные соединения
Биологическая часть. Антибактериальную активность определяли диско-диффузионным методом. Для исследований использовали плотную питательную среду для культивирования микроорганизмов ГРМ-агар ТУ 9398-020-78095326-2006. На приготовленный и разлитый в чашки Петри агар наносили 1-2 мл взвеси референтных штаммов Staphylococcus aureus ВКМ V-128 или Escherichia coli ВКМ V-820 густотой 5 единиц оптического бактериального стандарта мутности. Распределяли взвесь равномерно по поверхности среды, избыток удаляли. Чашки подсушивали 20 минут. В размеченные секторы помещали не-нагруженные препаратами диски из фильтр-картона. Разведение синтезированных препаратов готовили, добиваясь получения раствора вещества либо его мелкодисперсной суспензии, для чего растворяли препарат в 50 микролитрах диметилсульфоксида (ДМСО) и затем добавляли 5 мл горячей дистиллированной воды, тщательно перемешивали. При необходимости вещество растирали стеклянной палочкой и дополнительно подогревали. На диск наносили 15 микролитров полученной суспензии испытуемого соединения концентрацией 1000 мкг/мл, что составляет 15 микрограмм препарата на каждый диск. Подготовленные чашки помещали в термостат при 37°С на 24 часа. Препарат сравнения – фуразолидон. Оценивали величину зоны задержки роста бактериальной культуры вокруг диска в мм. Исследование протистоцидной активности проводили по методике на простейших вида Colpoda steinii (полевой изолят, коллекция лаборатории паразитологии ФГБНУ СКЗНИВИ). Работу выполняли в микропланшетах для постановки ИФА. В качестве среды для переживания простейших использовали смесь кипяченой водопроводной воды и стерильной дистиллированной воды в равных объемах. Препарат сравнения – байкокс (2,5%-ный раствор толтразурила). Результат оценивали по величине минимальной ингибирующей концентрации в мкг/мл. Исследование фунгистатической активности проводили методом диффузии в агар на культуре грибов вида Penicillium italicum ВКМ F-1279. На застывшую питательную среду №2 ГРМ (Сабуро) наносили 1 мл взвеси тест-культуры микромицета P. italicum (густотой 5 единиц оптического бактериального стандарта мутности). На диск наносили 15 микролитров суспензии испытуемого соединения на дистиллированной воде из расчёта 15 микрограммов препарата на каждый диск. Подготовленные чашки помещали в термостат при 26°С. Итоговый учет результатов производили через 72 часа. Препарат сравнения – фундазол. Активность оценивали в миллиметрах по величине зоны задержки роста культуры гриба вокруг диска. В процессе работы мы всегда используем зарекомендовавшие себя в течение долгого периода времени препараты сравнения, которые проявляют в работе качество, безопасность и эффективность в отношении исследуемых культур. Используемые препараты удобны тем, что всегда показывают стабильный результат. Результаты исследований и их обсуждение. При изучении биологической активности синтезированных соединений определили, что большинство синтезированных соединений обладают активностью в той или иной степени (табл. 3). Таблица 3 Биологическая активность синтезированных соединений
Изучение протистоцидной активности синтезированных соединений выявило высокие показатели у соединения №14, составившее 62,5±0,63 мкг/мл, что сопоставимо с показателями препарата сравнения – толтразурилом. Антибактериальная активность этого соединения выражена слабее, чем у препаратов сравнения и составляет 12±0,1 мм задержки роста в отношении Staphylococcus aureus VKM B-128 и 8±0,06 мм задержки роста в отношении Esherichia coli VKM B-820. Такой результат указывает на высокий потенциал соединения №14 для участия в разработке противопротозойных препаратов. Остальные соединения проявили умеренную и слабую про-тистоцидную активность и научного интереса в данном отношении не представляют. Наибольшую фунгистатическую активность наблюдали у соединения №10, она составила 15±0,12 мм задержки роста Penicillium italicum, что меньше, чем у препарата сравнения (фундазола). Но, поскольку положение 1 в молекуле №10 содержит липофильный алифатический заместитель (циклопентановое кольцо), то введение других подобных липофильных заместителей может стать перспективным направлением модификации структур данного ряда по усилению активности. Дальнейшие исследования в этом направлении считаем достаточно перспективными и интересными. Исследования антибактериальных свойств синтезированных соединения выявили максимальную активность в отношении Staphylococcus aureus VKM B-128 у соединения №1 – зона задержки роста составила 18±0,15 мм, это сопоставимо с результатами препарата сравнения – фуразолидоном, зона задержки роста которого составляет 18±0,14 мм. Чуть меньшую антибактериальную активность показали соединения №10 (15±0,12 мм) и №15 (15±0,13 мм). Соединение №15 показало наибольшую антибактериальную активность и в отношении Escherichia coli VKM B-820 – зона задержки роста составила 13±0,11 мм. Менее выраженная активность в отношении этого микроорганизма проявилась у соединений № 9 и №11 и составила 12±0,09 мм и 12±0,08 мм, соответственно. Соединение №9 интересно тем, что оно, единственное среди синтезированных, является более активным в отношении грамотрицательной палочки Escherichia coli, чем против Staphylococcus aureus. Это явление представляет научный интерес для дальнейших исследований механизма действия. Заключение. Ни одно соединение не проявило высокой активности одновременно по трем направлениям (протисто-цидная, фунгицидная и антибактериальная), что свидетельствует об узкой специфичной структуры соединений. Проведенные исследования по определению антибактериальной активности синтезированных соединений выявили селективность действия относительно грамположительных и грамо-трицательных бактерий. Высокая активность дикислоты №1 в отношении Staphylococcus aureus, сравнимая с активностью препарата сравнения фуразолидоном, а также заметная активность в отношении Esherichia coli дает основания считать перспективным синтез аналогов данной дикислоты как с донорными, так и акцепторными заместителями с целью усиления активности. Синтез и скрининг фталазинкарбоновых кислот является перспективным направлением поиска активно действующих субстанций с антибактериальной, анти-протозойной и фунгистатической активностью. Среди синтезированных соединений выделяется №14 высокой протисто-цидная активностью и №1 – антибактериальной активностью в отношении Staphylococcus aureus. Соединения №15 является самым сбалансированным антибактериальным веще- ством среди синтезированных; оно проявляет активность в отношении и Staphylococcus aureus, и Escherichia coli, что представляет интерес для дальнейшей модификации структуры с целью усиления активности. Список литературы: 1. Abdel-Ghany A. El-Helby, Helmy Sakr, Rezk R. Ayyad et al. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorganic Chemistry. 2020; (103): 104233. 2. Ahmed T.A. Boraei, Hanaa K. Ashour, El Sayed H. El Tamany et al. Design and synthesis of new phthalazine-based derivatives as potential EGFR inhibitors for the treatment of hepatocellular carcinoma. Bioorganic Chemistry. 2019; (85): 293-307. 3. Zubenko A. A., Sochnev V. S., Morkovnik A. S. et al. A new method for constructing pyrido[3’,2’:4,5]imidazo[1,2-b]pyridazine system and preparation of its derivatives. Mendeleev Communications. 2025; (35 (1): 24-26. 4. Angel H. Romero, Simon E. Lopez. In silico molecular docking studies of new potential 4-phthalazinyl-hydrazones on selected Trypanosoma cruzi and Leishmania enzyme targets. Journal of Molecular Graphics and Modelling. 2017; (76): 313-329. 5. Cinnathambi Subramani Maheswari, Rathinam Ramesh, Appaswami Lalitha. Antibacterial Evaluation of Some 3-Amino-1H-Pyrazolo[1,2-b] Phthalazine-2-Carboxamides by using Fish Scale Hydroxyapatite as a Heterogeneous Catalyst. Polycyclic Aromatic Compounds. 2021; (41 (10): 2221-2237. 6. Matiukhina A. K., Zorina-Tikhonova E. N., Gogoleva N. V. et al. Complexes of Fe(III), Co(III), and Cu(II) with Acylhydrazones Containing a Triphenylphosphonium Fragment: Synthesis, Crystal Structure, and Antibacterial Activity. Russian Journal of Coordination Chemistry. 2025; (51 (5): 328-339. 7. Kamilia M. Amin, Flora F. Barsoum, Fadi M. Awadallah, Nehal E. Mohamed. Identification of new potent phthalazine derivatives with VEGFR-2 and EGFR kinase inhibitory activity. European Journal of Medicinal Chemistry. 2016; (123): 191-201. 8. Mai M. Khalaf, Hany M. Abd El-Lateef, M. Gouda et al. New phthalazine based nickel (II), cobalt (II), and copper (II) mixed-ligand complexes; characterization, physicochemical properties, anti-inflammatory, antifungal, antibacterial, DFT and molecular docking exploration. Journal of the Indian Chemical Society. 2024; (101 (8): 101191. 9. Mohamed S. Behalo, Iman A. Gad El-karim, Randa Rafaat. Synthesis of Novel Phthalazine Derivatives as Potential Anticancer and Antioxidant Agents Based on 1-Chloro-4-(4-phenoxyphenyl)phthalazine. Journal of Heterocyclic Chemistry. 2017; (54 (6): 3591-3599. 10. Mohamed S. Nafie, Sherif Ashraf Fahmy, Shaima H. Kahwash et al. Recent advances on anticancer activity of benzodiazine heterocycles through kinase inhibition. RSC Advances. 2025; (15 (7): 5597-5638. 11. Mosaad S.M. Abdalla, Mohamed I. Hegab, Nageh A. Abo Taleb et al. Synthesis and anti-inflammatory evaluation of some condensed [4-(3,4-dimethylphenyl)-1(2H)-oxo-phthalazin-2-yl]acetic acid hydrazide. European Journal of Medicinal Chemistry. 2010; (45 (4): 1267-1277. 12. Bovkunova A. A., Bazhina E. S., Shmelev M. A. et al. New Manganese(II) Coordination Compounds with 4-((1H-Pyrrol-2-yl)methylene-amino)-4 H -1,2,4-triazole. Russian Journal of Coordination Chemistry. 2025; (51 (6): 472-487. 13. Prithwiraj De, Michel Baltas, Delphine Lamoral-Theys et al. Synthesis and anticancer activity evaluation of 2(4-alkoxyphenyl) cyclopropyl hydrazides and triazolo phthalazines. Bioorganic & Medicinal Chemistry. 2010; (18 (7): 2537-2548. 14. Sajjad Keshipour, Salman Shojaei, Ahmad Shaabani. A novel one-pot isocyanide-based four-component reaction: synthesis of highly functionalized 1H-pyrazolo[1,2-b]phthalazine-1,2-dicarboxylates and 1H-pyrazolo[1,2-a]pyridazine-1,2-dicarboxylates. Tetrahedron. 2012; (68 (31): 6141-6145. 15. Sara M. Emam, Samir M. El Rayes, Ibrahim A. I. Ali et al. Synthesis and Cytotoxicity of Novel ?-Ala-Phthalazine-Based Derivatives as VEGFR2 Inhibitors and Apoptosis-Inducers Against Liver Cancer. Polycyclic Aromatic Compounds. 2025; (45 (5): 771-792. 16. Sumera Zaib, Imtiaz Khan. Synthetic and medicinal chemistry of phthalazines: Recent developments, opportunities and challenges. Bioorganic Chemistry. 2020; (105): 104425. 17. Walid Ettahiri, Driss Fadili, Mohammed Mater Alanazi et al. Anticancer effect of novel phthalazines derivatives: Synthesis, biological evaluation, DFT, ADMET and docking study. Journal of Molecular Structure. 2025; (1327): 141173. 18. Yurong Yang, Fuling Xiao, Jianping Zuo et al. Structural optimization of phthalazine derivatives for anti-HBV activities to improve oral bioavailability. Bioorganic & Medicinal Chemistry. 2025; (128): 118259. Сведения об авторах: Зубенко Александр Александрович, доктор биологических наук, главный научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-928-6049743; e-mail: alexsandrzubenko@yandex.ru. Фетисов Леонид Николаевич, кандидат ветеринарных наук, ведущий научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-908-1978224; e-mail: fetisoff.leonid2018@yandex.ru. Яровая Наталья Александровна, лаборант-исследователь творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-999-2340481; e-mail: tkhim.sintez@yandex.ru. Байрамова Виолетта Николаевна, лаборант-исследователь творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-938-4043648; e-mail: tkhim.sintez@yandex.ru. Ответственный за переписку с редакцией: Святогорова Александра Евгеньевна, кандидат сельскохозяйственных наук, ведущий научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-988-9525755; e-mail: sviatogorova.a@yandex.ru. Заявленный вклад авторов: рукопись была написана благодаря вкладу всех авторов. Все авторы одобрили окончательную версию рукописи. Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
|


| 2011 © Ветеринария Кубани | Разработка сайта - Интернет-Имидж | |
|---|---|---|